The structural and dynamical properties of complex alloys are fundamentally controlled by the motion of atoms in a free energy landscape. Direct molecular dynamics simulations, however, cannot access the timescales required to sufficiently explore this landscape and describe how it changes due to diffusion or interface motion. Field-based methods offer a way forward, but strong approximations must be made in the free energy functionals that guide the dynamics. This project aims to develop a new classical density functional based approach (so-called alloy diffusional molecular dynamics) for studying solute redistribution at defects and interfaces in multicomponent alloys. The project builds on previous work at UBC that provides insight into the physics of solute diffusion due to the presence of atomistic, crystalline defects and the resulting structural changes in the material. This technique treats the atomistic topology of a crystalline material by density fields and evolves the alloy composition based on an approximated atomistic free energy functional and continuum description of mass transport. As a result, we can study diffusion processes that are inaccessible with particle based molecular dynamics simulations but retain crucial aspects of the discrete nature of matter.
 edge and (b) screw dislocation cores in Al-2.5%atMg alloy")
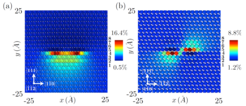
References:
E. Dontsova, J. Rottler, and C. W. Sinclair, Solute-defect interactions in Al-Mg alloys from diffusive variational Gaussian calculations, Phys. Rev. B 90, 174102 (2014)
E. Dontsova, J. Rottler, and C. W. Sinclair, Solute segregation kinetics and dislocation depinning in a binary alloy, Phys. Rev. B 91, 224103 (2015)
Principal Investigators:
Joerg Neugebauer (Max-Planck Institut fuer Eisenforschung)
Joerg Rottler (UBC) jrottler [at] phas.ubc.ca
Chad Sinclair (UBC)